Qué son las terapias ARN, revolución en el tratamiento de enfermedades raras
Están cambiando la vida de pacientes de enfermedades que hasta hace poco no eran tratables, pero el costo de las terapias es alto y muchos pacientes luchan para acceder a ellas.
Catalina, una niña de siete años que no podía alzar los brazos, ahora se peina sola.
Carlos, un cirujano que estaba condenado, en sus propias palabras, "a una muerte lenta y dolorosa", sigue operando y agradece una segunda oportunidad en la vida.
Ambos padecen enfermedades "raras", como se denomina a las que afectan a menos de una en dos mil personas.
Y en ambos casos, hasta hace poco, no había ni tratamiento ni esperanza.
El cambio en la vida de Catalina, Carlos y muchos otros pacientes se debe a la aparición en los últimos años de las llamadas "terapias ARN" (RNA en inglés).
"En mi opinión estamos viviendo una auténtica revolución silenciosa", dijo a BBC Mundo Rubén Artero, catedrático de genética de la Universidad de Valencia.
"Mientras durante toda la historia de la humanidad hemos dependido de productos naturales o pequeñas moléculas orgánicas para curar enfermedades, en los últimos años estamos desarrollando las herramientas para utilizar moléculas de RNA para controlar la expresión de los genes de una manera muy específica".
"Esto abre todo un abanico de posibilidades nuevas para atacar patologías previamente intratables".
Pero estos tratamientos revolucionarios tienen un alto costo. Y muchas familias como la de Santiago, un adolescente con la misma enfermedad de Catalina, siguen su lucha sin cuartel para acceder a los nuevos medicamentos.
En BBC Mundo te contamos qué son las terapias ARN o RNA, cómo funcionan en el caso de dos medicamentos, y por qué se han vuelto un eje central en la vida de miles de pacientes en el mundo como Carlos, Catalina y Santiago.
Qué son las terapias ARN
Tal vez hayas oído hablar de técnicas genéticas prometedoras como CRISPR/Cas, cuyas creadoras recibieron este año el premio Nobel de química.
Estas técnicas buscan editar o corregir directamente el ADN (DNA en inglés) dentro de los genes, pero las terapias ARN actúan de una manera diferente.

"Mientras en el DNA o ADN están todas las instrucciones genéticas de la célula, los RNAs son copias de cada una de esas instrucciones", explicó el profesor Artero.
La principal ventaja de actuar a nivel del ARN no es tanto lo que se consigue, como lo que se evita, agregó el experto.
"Mientras las técnicas basadas en CRISPR/Cas pueden cometer errores y potencialmente mutar genes críticos para el organismo sobre los que no deben actuar (por ejemplo sobre genes supresores de tumores), las técnicas que actúan a nivel del RNA no realizan cambios permanentes sobre el DNA y por tanto se consideran más seguras".
El papel del ARN
Hay distintos tipos de ARN y estas moléculas pueden cumplir funciones diferentes.
Una fundamental, en el caso del llamado ARN mensajero, es transportar una copia de instrucciones genéticas desde los genes en el núcleo de la célula hasta otro sitio fuera del núcleo, un complejo llamado ribosoma, donde las instrucciones son usadas para elaborar proteínas.
"Un RNA mensajero es una copia de la información contenida en un gen en una célula, pero en un formato que permite la síntesis de proteínas por parte del ribosoma", señaló Artero.
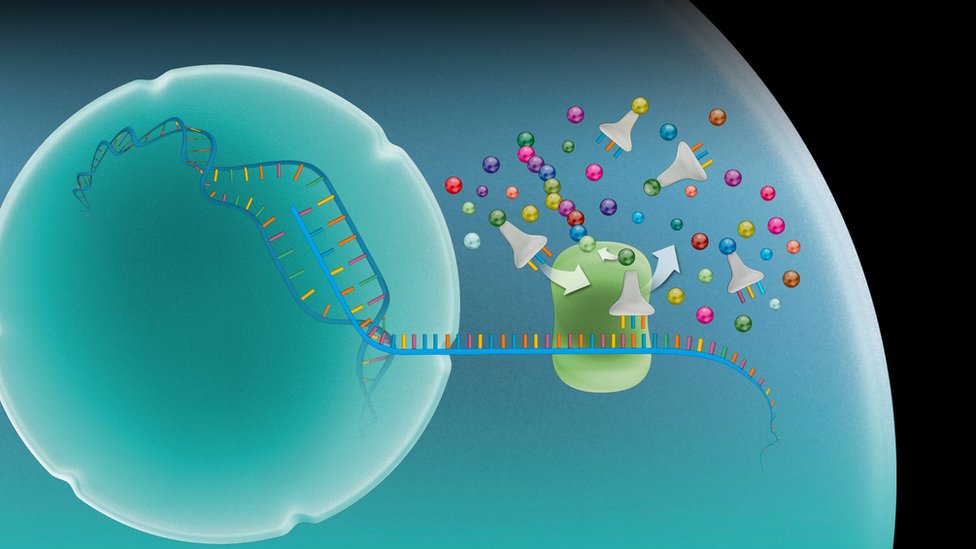
Algunas vacunas que se están desarrollando para covid-19 administran ARN mensajero. En este caso el ARN contiene instrucciones para que el organismo del paciente elabore parte de una proteína del virus del covid, y se despierte así una reacción inmune.
Las terapias ARN para enfermedades raras también pueden actuar sobre el ARN mensajero. Pero hay distintos tipos de terapias con estrategias diferentes.
Algunos medicamentos, por ejemplo, usan moléculas sintéticas de ARN que reconocen específicamente el ARN mensajero de un gen defectuoso y lo reprimen.
La idea principal es siempre que estas terapias no cambian a los genes directamente, sino a la expresión de esos genes.
La historia de Carlos
Un ejemplo de terapia ARN es Patisiran, un medicamento del laboratorio Alnylam de Estados Unidos que fue aprobado en 2018 en ese país y en Europa.
Patisiran es el primer medicamento efectivo para la amiloidosis, una enfermedad genética en la que versiones anormales de una proteína, llamada transtiretina o TTR, se depositan en tejidos, especialmente en nervios periféricos y en el corazón.
"Es una enfermedad degenerativa progresiva y letal", dijo a BBC Mundo el cirujano de manos español Carlos Heras-Palou, quien es oriundo de Mallorca pero vive y trabaja en Inglaterra hace más de dos décadas.
Heras-Palou fue diagnosticado en 2004 con un tipo hereditario de la enfermedad llamado amiloidosis hATTR, que afecta a unas diez mil personas en el mundo.
"Empecé a notar que me dolían mucho las manos y los pies, perdía la sensibilidad, me daban hormigueos y tenía dolor por la noche", relató.
El cirujano, quien tenía entonces 39 años y dos hijas pequeñas, inmediatamente sospechó cuál era la causa. Su madre y su abuelo habían fallecido por la misma enfermedad.
"El diagnóstico es devastador, es como una sentencia de muerte pero de una muerte lenta y dolorosa, y no es que te lo cuenten. Lo sabes porque lo has visto en casa".
"La gente en esta situación ha visto a sus padres y a sus abuelos tener la enfermedad, sabes que en unos años estarás sin sensibilidad, paralizado en una silla de ruedas".
El impacto de la terapia
Heras-Palou tuvo la fortuna de ser incluido en un ensayo clínico de Patisiran en 2013 .
"El impacto que ha tenido el tratamiento en mi vida es enorme. Si no hubiera sido por esto yo no estaría vivo".
"Cuando tienes un medicamento te cambia la visión completamente porque por primera vez te das cuenta que esto es tratable. Y aún más importante es que sabes que tus hijos y otros miembros de la familia si desarrollan la enfermedad van a tener un tratamiento".
En Reino Unido menos de 200 pacientes como Heras-Palou reciben a domicilio el tratamiento con Patisiran, que consiste en una infusión intravenosa cada tres semanas.
El tratamiento es pagado por el Servicio Nacional de Salud, cuesta cerca de medio millón de dólares por paciente por año y es de por vida.
Los laboratorios que elaboran las nuevas terapias aseguran que desarrollarlas lleva muchos años de inversiones e investigación, y que al tratarse de enfermedades raras el número de beneficiarios es bajo. Varios críticos disputan esos argumentos y creen que los medicamentos deberían ser más accesibles.
De lo que no cabe dudas es del efecto que pueden tener los tratamientos.
"Patisiran me ha permitido parar la evolución de la enfermedad y en muchos aspectos estoy incluso mejor que antes", señaló el cirujano.
"Había llegado un momento en que me costaba caminar. El dolor se me fue, tenía problema de digestión y eso se fue, y sigo trabajando".
La hermana menor del cirujano fue diagnosticada con la misma enfermedad en 2013 y recibe Patisiran en Mallorca.
"Ella había dejado de trabajar y se desmayaba todo el tiempo. Ahora tiene un hijo pequeño, trabaja full time y va al gimnasio cada día".
"Lo curioso de estos medicamentos es que son tan específicos que no tienen efectos secundarios".
Patisiran actúa a través de un mecanismo llamado ARN de interferencia, cuyos descubridores recibieron el Premio Nobel de Medicina en 2006.
El mecanismo permite degradar el ARN mensajero de un gen defectuoso. En el caso de la enfermedad de Heras-Palou, al "silenciar" el gen defectuoso se reduce dramáticamente la producción de proteína dañina que se deposita en los tejidos.
La esperanza de Spinraza
Otra terapia ARN es el medicamento Spinraza del laboratorio estadounidense Biogen, que fue aprobado en 2016 en Estados Unidos y en 2017 en Europa para el tratamiento de la atrofia muscular espinal o AME.
La enfermedad es la principal causa genética de muerte en bebés y se estima que en Estados Unidos, por ejemplo, afecta a al menos 10 mil niños y adultos.
El medicamento fue posible gracias a una técnica desarrollada por los equipos de dos investigadores, el uruguayo Adrian Krainer y el estadounidense Frank Bennett, que en 2019 ganaron por su descubrimiento el premio Breakthrough, conocido como el Oscar de la Ciencia.

El AME afecta las neuronas motoras o motoneuronas, que transportan información desde el sistema nervioso central hasta los músculos.
"En los pacientes con AME hay una disminución del nivel de la proteína SMN, por lo que las motoneuronas en la médula espinal no funcionan bien y se degeneran. A medida que desaparecen las motoneuronas, los músculos se debilitan y atrofian", explicó a BBC Mundo Krainer, experto del Laboratorio Cold Spring Harbor, una institución de investigación científica en Nueva York.
"El Spinraza logra que las motoneuronas vuelvan a fabricar una cantidad adecuada de la proteína SMN, lo que les permite funcionar efectivamente, y se frena o evita el proceso degenerativo", agregó Krainer.
Los pacientes de AME carecen del gen SMN1, que contiene las instrucciones para elaborar la proteína SMN. Sin embargo, esos pacientes tienen dos o más copias de otro gen muy similar, el SMN2.
Normalmente el gen SMN2 permite elaborar cantidades muy reducidas de proteína SMN, pero el Spinraza actúa en el proceso que genera el ARN mensajero de este gen y de esta manera logra aumentar el nivel de proteína funcional.
"Cuando Spinraza se administra antes de que aparezcan los síntomas, en bebés de días o semanas de edad, impide el inicio de la enfermedad. Si se administra a pacientes que ya tienen la enfermedad, puede frenar el proceso degenerativo", explicó Krainer a BBC Mundo.
Para llevar el medicamento a las motoneuronas "es necesario inyectar Spinraza por punción lumbar en el líquido cefaloraquídeo", señaló el científico.
"Su vida media es muy larga, por lo que los pacientes precisan sólo cuatro inyecciones anuales".
La historia de Catalina
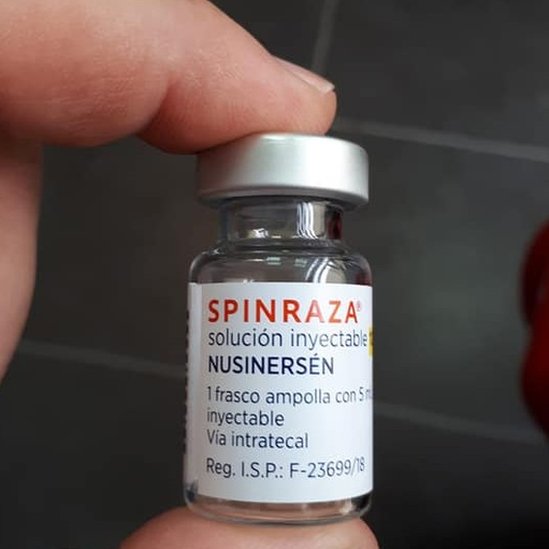
Spinraza es fabricado por el laboratorio Biogen de Estados Unidos y cada inyección cuesta en ese país 125 mil dólares.
En Uruguay se estima que hay cerca de 35 enfermos de AME, en su gran mayoría niños, pero hasta ahora solo 13 pacientes reciben Spinraza, que fue registrado en el país a un precio de 105 mil dólares cada dosis.
Para acceder al medicamento fue necesario recurrir a la vía judicial contra el Estado. Los juicios por cada niño fueron posibles gracias al consultorio jurídico gratuito de la Universidad de la República en Montevideo, en el que el abogado Juan Ceretta y otros docentes trabajan con cientos de estudiantes.

Uno de los fallos favorables fue para Catalina, que recibe el medicamento desde julio del año pasado.
"Cata puede peinarse, atarse el pelo con una gomita, jugar a maquillarse, ponerse un buzo, servirse un vaso de agua. Y antes no lograba ni siquiera mantener los brazos en alto", relató a BBC Mundo Federico Fuentes, el padre de Catalina.
"Ella tiene ahora punto de comparación 'para mejor', ya que ella notaba antes de recibir Spinraza su involución día a día y se frustraba o preguntaba por qué no podía hacer algo que ayer sí podía".
"A raíz de la medicación se la nota MUY FELIZ… ya que todo el tiempo está diciéndote, viste que ahora puedo hacer esto y también aquello ... y finaliza siempre con una hermosa sonrisa cada frase".

Krainer asegura que casos como el de Catalina son una fuente constante de inspiración.
"Es lo que me produce mayor satisfacción. O sea, ver que el esfuerzo conjunto de nuestro equipo y nuestros colaboradores durante tantos años, y el traslado de investigación básica, hayan culminado en un medicamento que le salva la vida a tantos niños y mejora la calidad de vida de miles de pacientes".
La historia de Santiago
El padre de Santiago, Pablo Correa, es uno de los fundadores de FAME Uruguay (Familias con AME) y se ha transformado en un referente para los pacientes y sus familiares.
Su hijo fue diagnosticado cuando era bebé.
"Lo que nota un papá cuando su hijo tiene AME es flacidez, es como un muñequito de trapo. Nosotros notamos que lo aupábamos y las piernas quedaban flácidas totalmente", señaló Correa a BBC Mundo.
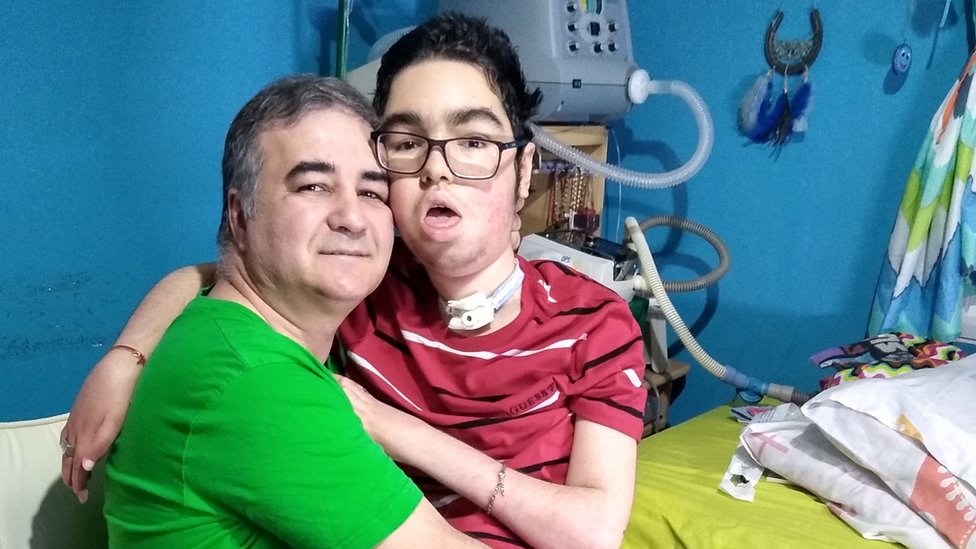
El niño fue diagnosticado con AME tipo 1, la forma más severa de la enfermedad, y los médicos le dieron no más de un año de vida.
"Nos dijeron que no había nada, nos dijeron llévelo para su casa y espere el desenlace de la vida. Pero Santi con los ojos nos decía algo".
"Nunca nos quedamos con que no hay nada. A los nueve meses hubo que ventilarlo. Hoy Santi lleva 17 años conectado a un respirador y nunca tuvo una infección respiratoria".
Desde que Santi fue diagnosticado su madre Silvia está dedicada por completo a cuidarlo. Y su padre, que es conductor de autobuses en Montevideo, no afloja en la búsqueda de un tratamiento para su hijo.
Ha acudido a todo tipo de congresos y no ha faltado a las reuniones de FAME Argentina. Correa recuerda la emoción que sintió cuando conoció en una de esas reuniones a Krainer.
"Estaba en primera fila cuando Krainer dijo que había inventado algo que iba a funcionar".

La lucha por Spinraza
Correa hizo el primer contacto desde Uruguay con Biogen y consiguió que Spinraza entrara al país a un precio menor que en Estados Unidos.
Pero no ha logrado aún que su hijo acceda al medicamento.
"Para poder ir a juicio tenés que tener una prescripción médica. Y los médicos nos vienen diciendo que Santi ya superó la expectativa de vida, que el costo beneficio va a ser muy poco", afirmó.
"Venimos peleando contra la corriente, Santi viene demostrando que se puede, cumple 18 años el 28 de febrero y ya no podemos seguir hablando de expectativa de vida".
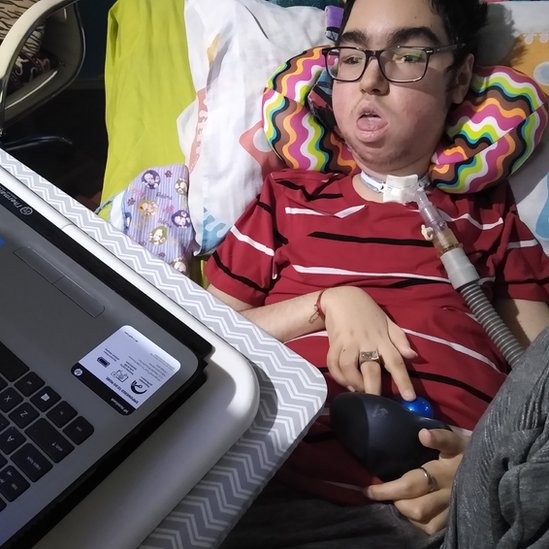
El adolescente usa una silla motorizada que controla con un dedo.
Tiene una profesora a domicilio y aprendió inglés "googleando y en youtube".
"A Santi le gusta mucho la computadora, es muy inteligente, usa un trackball, un ratón con una bolita. Con el diafragma golpea el ratoncito y escribe. Y sus juegos de computadora favoritos son los juegos de rol de abogacía".
La gran esperanza de la familia es que Santi y todos los enfermos de AME en el país accedan a Spinraza.
Los familiares piden al gobierno uruguayo que se involucre y negocie a nivel de país con el laboratorio, al igual que Argentina, que anunció en julio de este año un precio tope para cada inyección de US$27 mil.
"Es lo que venimos planteando, que Uruguay hable con el laboratorio y haga un plan de inclusión. El precio se puede bajar, otros países lo hicieron. Bajando el costo podemos acceder todos", afirmó Correa.
Consultado por BBC Mundo, el Ministro de Salud de Uruguay, Dr. Daniel Salinas, señaló por mail a través de su departamento de comunicaciones que "el grupo negociador está trabajando en un acuerdo país por este medicamento".

Una esperanza y un desafío
Los avances en terapias ARN continúan, con la búsqueda de tratamientos más fáciles de administrar y medicamentos nuevos para otras enfermedades.
Para Carlos Heras-Palou, el éxito de medicamentos como Patisiran "demuestra el principio de que estas terapias funcionan, y hay más de seis mil enfermedades raras, muchas de ellas genéticas, que tal vez en el futuro podrán tener tratamientos".
Pero acceder a esos medicamentos seguirá siendo en muchos casos un desafío.
Heras-Palou preside la asociación de pacientes de Amiloidosis hTTR en Reino Unido y está en contacto con asociaciones similares en España y América Latina.
"Vale la pena estar organizado, estar informado, tener una asociación de pacientes", aconseja.

El profesor Rubén Artero señaló que "debería asegurarse el acceso a terapias RNA por parte de los sistemas nacionales de salud y las aseguradoras privadas, aunque no cabe duda de que esto plantea un importante reto financiero, al menos a medio plazo".
Para Pablo Correa, el padre de Santi, las terapias ARN siguen siendo una aspiración.
"Le prometí a Santi hace 17 años que iba a tratar de hacer lo mejor posible para lograr algo para él. Hoy hay algo para él", afirmó a BBC Mundo.
"En otras partes del mundo sucede que un chico con 17 años reciba Spinraza, ¿por qué en Uruguay no?".
Recuerda que puedes recibir notificaciones de BBC Mundo. Descarga la nueva versión de nuestra app y actívalas para no perderte nuestro mejor contenido.